
Selected Publications
72 total, 33 first/co-first (†)/corresponding author (*)
Computational Chemistry and Machine Learning-Assisted Screening of Supported Amorphous Metal Oxide Nanoclusters for Methane Activation
Xijun Wang, Kaihang Shi, Anyang Peng, Randall Q. Snurr*
ACS Catal. 2024, 14, 18708–18721
Abstract:
Activating the C–H bond in methane represents a cornerstone challenge in catalytic research. While several supported metal oxide nanoclusters (MeO-NCs) have shown promise for this reaction, their optimal composition remains underexplored primarily due to the large number of possible compositions and their amorphous nature. This study addresses these challenges using computational approaches. Leveraging density functional theory (DFT) calculations, we began with a previously studied supported tetra-copper oxide nanocluster and systematically substituted its Cu sites with first-row transition metals (Mn, Fe, Co, Ni, and Zn). This process allowed us to examine the catalytic activity of 162 MeO-NCs with a variety of geometric and electronic structures, leading to 12 new compositions that outperformed the base nanocluster. Exploring the structure–activity relationships with machine learning, our analysis uncovered correlations between the intrinsic electronic and structural properties of the nanoclusters and the free energy barriers for methane activation despite the challenges posed by the structural flexibility of these amorphous nanoclusters. The results offer insights into the optimization of MeO-NCs for methane activation. Additionally, we developed a clustering model capable of distinguishing high-performing nanoclusters from less effective ones with strong tolerance to the interference from the structural flexibility of these amorphous nanoclusters. These findings help narrow down the material design space for more time-consuming high-level quantum chemical calculations, offering a promising pathway toward advancing eco-friendly methane conversion.

Probing the structure–property relationships of supported copper oxide nanoclusters for methane activation
Xijun Wang, Kaihang Shi, Anyang Peng, Randall Q. Snurr*
EES Catal. 2024, 2 (1), 351-364
Abstract:
Supported metal oxide nanoclusters (MeO-NCs) have gained significant attention for their remarkable versatility in various energy and sustainability applications. Despite rapid advancements in atomic-scale synthesis and characterization techniques, the rational design of MeO-NCs with desired catalytic properties remains challenging. This challenge arises from the elusive and difficult-to-quantify structure-catalytic property relationships, particularly in the case of amorphous nanoclusters. Exploiting first-principles calculations at the density functional theory (DFT) level, we conducted a systematic investigation into the growth, geometries, and catalytic performance of a series of tetra-copper oxide nanoclusters (Cu4O-NCs) for methane activation. Focusing on the most representative geometries, we applied machine learning to extract two physically insightful descriptors involving the spin density, the p-band center of the oxygen site, and the d-band center of adjacent Cu sites. These descriptors enable us to predict free energy barriers associated with both the homolytic and heterolytic mechanisms of methane activation. This descriptor-driven approach enables rapid and intuitive prediction of the preferred reaction mechanism. Our findings lay a solid foundation for future advancements in catalysts based on amorphous nanoclusters and provide valuable insights into the mechanistic landscape of methane activation.

Fine-Tuning Dual Single-Atom Metal Sites on Graphene toward Enhanced Oxygen Reduction Reaction Activity
Evan Xie, Xijun Wang*
J. Phys. Chem. Lett. 2023, 14, 42, 9392–9402
Abstract:
The oxygen reduction reaction (ORR) remains at the forefront of research in diverse energy and sustainability domains. While graphene-supported single-atom catalysts (SACs) have garnered attention for optimizing ORR efficiency, tailoring the interactions between adjacent single-atom sites presents intricate challenges. In this study, we leveraged density functional theory (DFT) calculations and cutting-edge machine learning (ML) techniques to explore 144 graphene-supported SACs, featuring interacting M1–N4 and M2–N4 moieties (M1, M2 = Mn, Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag Ir, Pt, Au), denoted as M1–M2. By tailoring these interactions, we discovered 13 exceptional SACs outperforming the benchmark catalyst Fe(OH)–N4, including the best-performing Fe–Pd and several non-noble-metal SACs like Fe–Ag, Ag–Cu, and Ag–Ag. Venturing further, our ML models effectively capture the correlation between single-atom metal properties and overpotential, offering tools for rational electrocatalyst design. Our study illuminates the path to efficient SAC-catalyzed ORR, fostering a sustainable, energy-efficient future.

Quantitatively Determining Surface−Adsorbate Properties from Vibrational Spectroscopy with Interpretable Machine Learning
Xijun Wang, Shuang Jiang, Wei Hu, Sheng Ye, Tairan Wang, Fan Wu, Li Yang, Xiyu Li, Guozhen Zhang, Xin Chen,* Jun Jiang,* Yi Luo*
J. Am. Chem. Soc. 2022, 144, 35, 16069–16076
Abstract:
Learning microscopic properties of a material from its macroscopic measurables is a grand and challenging goal in physical science. Conventional wisdom is to first identify material structures exploiting characterization tool, such as spectroscopy, and then to infer properties of interest, often with assistance of theory and simulations. This indirect approach is pestered by accumulation of errors from retrieving structures from spectral signals and the lack of quantitative structure-property relationship. A new pathway directly from spectral signals to microscopic properties is highly desirable, as it would offer valuable guidance towards materials evaluation and design via spectroscopic measurements. Herein, we exploit machine learned vibrational spectroscopy to establish quantitative spectrum-property relationships. Key interaction properties of substrate-adsorbate systems, including adsorption energy and charge transfer, are quantitatively determined directly from Infrared and Raman spectroscopic signals of the adsorbates. The machine learned spectrum-property relationships are presented as mathematical formulas, which are physically interpretable and therefore transferrable to a series of metal/alloy surfaces. The demonstrated ability of quantitative determining hard-to-measure microscopic properties using machine learned spectroscopy will significantly broaden the applicability of conventional spectroscopic techniques for materials design and high throughput screening under operando conditions.

High-Throughput Oxygen Chemical Potential Engineering of Perovskite Oxides for Chemical Looping Applications
Xijun Wang, Yunfei Gao, Emily Krzystowczyk, Sherafghan Iftikhar, Jian Dou, Runxia Cai, Haiying Wang, Chongyan Ruan, Sheng Ye, Fanxing Li*
Energy Environ. Sci. 2022, 15, 1512-1528
Abstract:
Chemical looping (CL) represents a versatile, emerging strategy for sustainable chemical and energy conversion. Designing metal oxide oxygen carriers with suitable redox properties remains one of the most critical challenges to CL due to the considerably different thermodynamic property requirements for different applications. Taking SrFeO3-δ as a base-structure, this study seeks to rationally substitute its A- and/or B-site cations to tailor the equilibrium oxygen partial pressure over 20 orders of magnitude. 2,401 SrxA1-xFeyB1-yO3-δ perovskite-phase structures were investigated using high-throughput density functional theory (DFT) and 227,273 high-entropy perovskites were screened via machine learning (ML). This significantly expands the materials design space. While most of the compositions predicted are new and nonobvious, 19 previously reported oxygen carriers, with excellent redox properties, were correctly identified by the algorithm. Moreover, we experimentally demonstrated 15 new oxygen carriers with superior redox performance. These results support the effectiveness of the high-throughput approaches for accelerated materials discovery.

Electric Dipole Descriptor for Machine Learning Prediction of Catalyst Surface–Molecular Adsorbate Interactions
Xijun Wang, Sheng Ye, Wei Hu, Edward Sharman, Ran Liu, Yan Liu, Yi Luo, Jun Jiang*
J. Am. Chem. Soc. 2020, 142, 17, 7737–7743
Abstract:
The challenge of evaluating catalyst surface–molecular adsorbate interactions holds the key for rational design of catalysts. Finding an experimentally measurable and theoretically computable descriptor for evaluating surface–adsorbate interactions is a significant step toward achieving this goal. Here we show that the electric dipole moment can serve as a convenient yet accurate descriptor for establishing structure–property relationships for molecular adsorbates on metal catalyst surfaces. By training a machine learning neural network with a large data set of first-principles calculations, we achieve quick and accurate predictions of molecular adsorption energy and transferred charge. The training model using NO/CO@Au(111) can be extended to study additional substrates such as Au(001) or Ag(111), thus exhibiting extraordinary transferability. These findings validate the effectiveness of the electric dipole descriptor, providing an efficient modality for future catalyst design.

Isolating hydrogen from oxygen in photocatalytic water splitting with a carbon-quantum-dot/carbon-nitride hybrid
Xijun Wang, Xiang Jiang, Edward Sharman, Li Yang, Xiyu Li, Guozhen Zhang, Jin Zhao, Yi Luo, Jun Jiang*
J. Mater. Chem. A 2019, 7, 6143-6148
Abstract:
The practical utilization of solar-driven water splitting is restricted by the difficulty of this type of splitting in producing hydrogen and oxygen molecules with the same photocatalyst. For the separate delivery of photo-generated electron and hole carries to the reduction and oxidation sites in photocatalysts, close reduction–oxidation distances are required, which, however, promote reverse reactions or even damaging explosions due to the mixing of hydrogen and oxygen-containing products. To overcome this challenge, we designed a hybrid structure of carbon-quantum-dots (CQDs) attached to a single-layered carbon nitride (C3N) material. Using first-principles calculations, we showed that the hybrid can harvest visible and infrared light for water splitting. It holds very close reduction–oxidation sites, ensuring the rapid delivery of photo-generated electrons and holes to the inner CQD and outer C3N layers separately. The holes then break the water molecules and produce protons at the outer C3N. Due to electrostatic attraction, the protons penetrate through C3N to react with the photo-generated electrons on CQD and produce hydrogen molecules. Since neither oxygen species nor hydrogen molecules can migrate through the C3N layer, hydrogen products are completely isolated from the oxygen species; this inhibits reverse reactions. These metal-free hybrids are thus appealing photocatalysts to integrate very close yet well-separated reduction–oxidation sites for practical solar energy conversion and hydrogen energy utilization.

Metal-enhanced hydrogenation of graphene with atomic pattern
Xijun Wang, Guozhen Zhang, Zhaowu Wang, Li Yang, Xiyu Li, Jun Jiang,* Yi Luo*
Carbon 2019, 143, 700–705
Abstract:
Graphene hydrogenation is an attractive approach to functionalize graphene. However, the hydrogenating treatment could only be achieved under harsh conditions due to the weak hydrogen affinity of graphene. It has also made the direct writing of electronic circuits on graphene by hydrogenation very difficult. Here we propose a metal-enhanced approach to hydrogenate graphene with atomic pattern that enables to produce hydrogenation pattern on demand. First-principles calculations reveal that certain metals (Cu, Ag, Al) attached to graphene can significantly lower the energy barrier for hydrogen binding process, while the hydrogen binding energy itself is much enhanced. Such metal-promoted hydrogenation is spatially localized, which paves the way to precisely write pre-designed hydrogenation patterns on graphene with well-controlled metallic clusters and tips.

Material descriptors for photocatalyst/catalyst design
Xijun Wang, Guozhen Zhang, Li Yang, Edward Sharman, Jun Jiang*
WIREs Comput. Mol. Sci. 2018, e1369
Abstract:
Rational design of high‐performance photocatalysts/catalysts is crucial for sustainable development. To achieve this goal, a comprehensive understanding and precise description of structure–performance relationships of photocatalysts/catalysts are highly desirable. While photocatalysis/catalysis involves complex systems and processes, approximate descriptors have been proposed for sorting out simple pictures of complicated structure–performance relationships concerned. In this review, some important descriptors involved in photocatalyst/catalyst design including work function, dipole moment, d‐band center, and Fermi softness are reviewed first with special attention being paid to their working mechanisms and applications. Then strategies of tuning photocatalytic/catalytic performance on the basis of these descriptors are outlined. Finally, challenges and opportunities for photocatalyst/catalyst design based on descriptor control are discussed.

Insight into Electronic and Structural Reorganizations for Defect-Induced VO2 Metal–Insulator Transition
Xijun Wang, Zhaowu Wang, Guozhen Zhang, Jun Jiang*
J. Phys. Chem. Lett. 2017, 8, 13, 3129–3132
Abstract:
An oxygen vacancy defect in monoclinic VO2 has been shown to modulate the metal–insulator transition (MIT) at room temperature. However, as the electronic and structural reorganizations occur simultaneously, the origin of MIT is still unclear. Here we performed first-principles calculations to examine electronic variations separately from structural reorganizations during MIT. It was found that the oxygen defect induces electronic reorganization by creating polarized 3d orbitial electrons, while structure reorganization makes the conduction band edge states available for occupation. The conduction band states thus hold polarized charges that delocalize over space, bestowing metallic property on the originally insulated VO2. A linear relationship for the number of polarized electrons and the defect concentration is revealed, which would lead to cost-effective control of VO2 MIT behavior by defect engineering.

Net Electronic Charge as an Effective Electronic Descriptor for Oxygen Release and Transport Properties of SrFeO3-based Oxygen Sorbents
Xijun Wang, Emily Krzystowczyk, Jian Dou, Fanxing Li*
Chem. Mater. 2021, 33, 7, 2446–2456
Abstract:
Perovskite oxides, as oxygen sorbents, exhibit excellent potential in thermochemical redox applications such as chemical looping air separation (CLAS), resulting from their excellent redox properties and high tunability. The structural and compositional flexibility of perovskites, while making these mixed oxides highly versatile, also poses challenges for their design and optimization. Moreover, the elevated operating temperature and dynamic oxygen partial pressure swings during redox processes add additional complications for oxygen sorbent design. From a fundamental standpoint, finding simple yet effective descriptors for the effect of cation substitutions on oxygen sorbents’ redox and oxygen transport properties are of significant importance for rational optimization of perovskite-based oxygen sorbents. In the present study, a series of A/B-site-doped SrFeO3‑δ perovskites are investigated using density functional theory (DFT) + U simulations. From these simulations, an effective electronic structural descriptor, the net electronic charge (Δe) of oxygen anions, is extracted based on its excellent correlations with vacancy formation energy, experimentally derived oxygen capacity, and oxygen vacancy migration barrier. These findings provide an effective tool to correlate the oxygen sorbents’ redox performances with a simple yet physically meaningful descriptor for rational design and optimization of perovskites in the context of CLAS.

Carbon Monoxide Oxidation Promoted by Surface Polarization Charges in a CuO/Ag Hybrid Catalyst
Xijun Wang, Chuanyi Jia, Edward Sharman, Guozhen Zhang, Xin Li,* Jun Jiang
Sci. Rep. 2020, 10, 1, 1-9
Abstract:
Composite structures have been widely utilized to improve material performance. Here we report a semiconductor-metal hybrid structure (CuO/Ag) for CO oxidation that possesses very promising activity. Our first-principles calculations demonstrate that the significant improvement in this system’s catalytic performance mainly comes from the polarized charge injection that results from the Schottky barrier formed at the CuO/Ag interface due to the work function differential there. Moreover, we propose a synergistic mechanism underlying the recovery process of this catalyst, which could significantly promote the recovery of oxygen vacancy created via the M-vK mechanism. These findings provide a new strategy for designing high performance heterogeneous catalysts.

Bandgap tuning of C3N monolayer: A first-principles study
Liyan Xie, Li Yang, Wanying Ge, Xijun Wang,* Jun Jiang
Chem. Phys. 2019, 520, 1, 40–46
Abstract:
TheThe newly found graphene-like material C3N exhibits great potential in a variety of important applications, due to its unique topological and electronic structures. To extend the utilization, a crucial challenge is to make its intrinsic bandgap (1.03 eV) tunable. Here we performed first-principles calculations to investigate the band structure variations of C3N monolayer under various surface modification treatments, including defect engineering, surface decoration and substitutional doping. Results show that those treatments can induce impurity states, orbital rehybridization, and n- or p-type doping simultaneously, and therefore enable effective band structure adjustment. Importantly, some linear relationships between the bandgap and doping concentration are revealed, paving the way for precise control of C3N bandgap.

Structure–Activity Relationship Insights for Organophosphonate Hydrolysis at Ti(IV) Active Sites in Metal–Organic Frameworks
Mohammad Rasel Mian,† Xijun Wang,† Xingjie Wang, Kent O. Kirlikovali, Haomiao Xie, Kaikai Ma, Kira M. Fahy, Haoyuan Chen, Timur Islamoglu*, Randall Q. Snurr*, and Omar K. Farha*
J. Am. Chem. Soc. 2023, 145, 13, 7435–7445
Abstract:
Organophosphorus nerve agents are among the most toxic chemicals known and remain threats to humans due to their continued use despite international bans. Metal–organic frameworks (MOFs) have emerged as a class of heterogeneous catalysts with tunable structures that are capable of rapidly detoxifying these chemicals via hydrolysis at Lewis acidic active sites on the metal nodes. To date, the majority of studies in this field have focused on zirconium-based MOFs (Zr-MOFs) that contain hexanuclear Zr(IV) clusters, despite the large toolbox of Lewis acidic transition metal ions that are available to construct MOFs with similar catalytic properties. In particular, very few reports have disclosed the use of a Ti-based MOF (Ti-MOF) as a catalyst for this transformation even though Ti(IV) is a stronger Lewis acid than Zr(IV). In this work, we explored five Ti-MOFs (Ti-MFU-4l, NU-1012-NDC, MIL-125, Ti-MIL-101, MIL-177(LT), and MIL-177(HT)) that each contains Ti(IV) ions in unique coordination environments, including monometallic, bimetallic, octanuclear, triangular clusters, and extended chains, as catalysts to explore how both different node structures and different linkers (e.g., azolate and carboxylate) influence the binding and subsequent hydrolysis of an organophosphorus nerve agent simulant at Ti(IV)-based active sites in basic aqueous solutions. Experimental and theoretical studies confirm that Ti-MFU-4l, which contains monometallic Ti(IV)–OH species, exhibits the best catalytic performance among this series with a half-life of roughly 2 min. This places Ti-MFU-4l as one of the best nerve agent hydrolysis catalysts of any MOF reported to date.

Accelerated Perovskite Oxide Development for Thermochemical Energy Storage by a High-Throughput Combinatorial Approach
Runxia Cai,† Hilal Bektas,† Xijun Wang,† Kyle McClintock, Lauren Teague, Kunran Yang, Fanxing Li*
Adv. Energy Mater. 2023, 2203833
Abstract:
The structural and compositional flexibility of perovskite oxides and their complex yet tunable redox properties offer unique optimization opportunities for thermochemical energy storage (TCES). To improve the relatively inefficient and empirical-based approaches, a high-throughput combinatorial approach for accelerated development and optimization of perovskite oxides for TCES is reported here. Specifically, thermodynamic-based screening criteria are applied to the high-throughput density functional theory (DFT) simulation results of over 2000 A/B-site doped SrFeO3-δ. 61 promising TCES candidates are selected based on the DFT prediction. Of these, 45 materials with pure perovskite phases are thoroughly evaluated. The experimental results support the effectiveness of the high-throughput approach in determining both the oxygen capacity and the oxidation enthalpy of the perovskite oxides. Many of the screened materials exhibit promising performance under practical operating conditions: Sr0.875Ba0.125FeO3-δ exhibits a chemical energy storage density of 85 kJ kgABO3-1 under an isobaric condition (with air) between 400 and 800 °C whereas Sr0.125Ca0.875Fe0.25Mn0.75O3-δ demonstrates an energy density of 157 kJ kgABO3-1 between 400 °C/0.2 atm O2 and 1100 °C/0.01 atm O2. An improved set of optimization criteria is also developed, based on a combination of DFT and experimental results, to improve the effectiveness for accelerated development of redox-active perovskite oxides.

Alkali metal halide–coated perovskite redox catalysts for anaerobic oxidative dehydrogenation of n-butane
Yunfei Gao,† Xijun Wang,† Noel Corolla, Tim Eldred, Arnab Bose, Wenpei Gao, Fanxing Li*
Sci. Adv. 2022, 8, eabo7343
Abstract:
Oxidative dehydrogenation (ODH) of n-butane has the potential to efficiently produce butadiene without equilibrium limitation or coke formation. Despite extensive research efforts, single-pass butadiene yields are limited to

Selective catalytic oxidation of ammonia to nitric oxide via chemical looping
Chongyan Ruan,† Xijun Wang,† Chaojie Wang, Lirong Zheng, Lin Li, Jian Lin, Xiaoyan Liu, Fanxing Li,* Xiaodong Wang*
Nat. Commun. 2022, 13, 718
Abstract:
Selective oxidation of ammonia to nitric oxide over platinum-group metal alloy gauzes is the crucial step for nitric acid production, a century-old yet greenhouse gas and capital intensive process. Therefore, developing alternative ammonia oxidation technologies with low environmental impacts and reduced catalyst cost are of significant importance. Herein, we propose and demonstrate a chemical looping ammonia oxidation catalyst and process to replace the costly noble metal catalysts and to reduce greenhouse gas emission. The proposed process exhibit near complete NH3 conversion and exceptional NO selectivity with negligible N2O production, using nonprecious V2O5 redox catalyst at 650 oC. Operando spectroscopy techniques and density functional theory calculations point towards a modified, temporally separated Mars-van Krevelen mechanism featuring a reversible V5+/V4+ redox cycle. The V = O sites are suggested to be the catalytically active center leading to the formation of the oxidation products. Meanwhile, both V=O and doubly coordinated oxygen participate in the hydrogen transfer process. The outstanding performance originates from the low activation energies for the successive hydrogen abstraction, facile NO formation as well as the easy regeneration of V=O species. Our results highlight a transformational process in extending the chemical looping strategy to producing base chemicals in a sustainable and cost-effective manner.

An Adjacent Atomic Platinum Site Enables Single-Atom Iron with High Oxygen Reduction Reaction Performance
Ali Han,† Xijun Wang,† Kun Tang, Zedong Zhang, Chenliang Ye, Kejian Kong, Haibo Hu, Lirong Zheng, Peng Jiang, Changxin Zhao, Qiang Zhang, Dingsheng Wang,* Yadong Li
Angew. Chem. Int. Ed. 2021, 60, 19262-19271
Abstract:
Single-atoModulation effect has been widely investigated to tune the electronic state of single-atomic M-N-C catalysts to enhance the activity of oxygen reduction reaction (ORR). However, the in-depth study of modulation effect is rarely reported for the isolated dual-atomic metal sites. Interestingly, guided by the first-principles simulations, we find that the catalytic activities of Fe-N4 moiety can be enhanced by the adjacent Pt-N4 moiety through the modulation effect, in which the Pt-N4 acts as the modulator to tune the 3d electronic orbitals of Fe-N4 active site and optimize ORR activity. Inspired by this principle, we design and synthesize the electrocatalyst that comprises isolated Fe-N4/Pt-N4 moieties dispersed in the nitrogen-doped carbon matrix (Fe-N4/Pt-N4@NC) and exhibits a half-wave potential of 0.93 V vs. RHE and negligible activity degradation (∆E1/2 = 8 mV) after 10000 cycles in 0.1 M KOH. We also demonstrate that the modulation effect is not effective for optimizing the ORR performances of Co-N4/Pt-N4 and Mn-N4/Pt-N4 systems. These results have refreshed the knowledge of adjacent dual metal sites at the atomic-level and provided rational guidance for the design of efficient electrocatalysts.

One-Step Synthesis of Single-Site Vanadium Substitution in 1T-WS2 Monolayers for Enhanced Hydrogen Evolution Catalysis
Ali Han,† Xiaofeng Zhou,† Xijun Wang,† Sheng Liu, Qihua Xiong, Wenjing Zhang, Fanxing Li, Dingsheng Wang,* Lain-Jong Li,* Yadong Li*
Nat. Commun. 2021, 12, 709
Abstract:
Metallic tungsten disulfide (WS2) monolayers have been demonstrated as promising electrocatalysts for hydrogen evolution reaction (HER) induced by the high intrinsic conductivity, however, the key challenges to maximize the catalytic activity are achieving the metallic WS2 with high concentration and increasing the density of the active sites. In this work, single-atom-V catalysts (V SACs) substitutions in 1T-WS2 monolayers (ca. 91% phase purity) are fabricated to significantly enhance the HER performance via a one-step chemical vapor deposition strategy. Atomic-resolution scanning transmission electron microscopy (STEM) imaging together with Raman spectroscopy confirm the atomic dispersion of V species on the 1T-WS2 monolayers instead of energetically favorable 2H-WS2 monolayers. The growth mechanism of V SACs@1T-WS2 monolayers is experimentally and theoretically demonstrated and reveals that the 1T-VS2 intermediates are found to critically determine the metallic WS2 1T phase growth. Benefiting from the high loading of single-atom V (4.0 at%), the HER activity of the intrinsic 1T-WS2 monolayers is significantly enhanced. Density functional theory (DFT) calculations demonstrate that the activated V-atom sites play vital important role in enhancing the HER activity. This work opens novel path to directly synthesize atomically dispersed single-metal catalysts on metastable materials as efficient and robust electrocatalysts.

Edge-Effect Enhanced Catalytic CO Oxidation by Atomically Dispersed Pt on Nitride-Graphene
Chuanyi Jia,† Xijun Wang,† Huabing Yin, Wenhui Zhong, Edward Sharman, Yi Luo, Jun Jiang*
J. Mater. Chem. A 2021, 9, 4, 2093-2098.
Abstract:
Single-atom catalysis has been of intense interest in recent years. Here, using first-principles simulations, we report a unique approach that obtains promising information about O2 activation and CO oxidation on active Pt-single-atom catalysts (Pt-SACs) on N-doped graphene (NGr). It is found that the activity of Pt-SACs can be tuned by changing the Pt-loading sites on Gr/NGr, with Pt on the zigzag edge of NGr having optimal performance. Employing charge analysis of various anchoring sites, we revealed that the catalytic performance has a strong dependence on the polarization charge on the Pt atoms. The correlations of those charges with binding energies and reaction barriers exhibit volcano-like trends. Polarization charge thus provides a parameter that allows to predict catalytic properties for different active sites, casting insights into electronic structure modulation of SACs on graphene-like supports.

Sharp-Tip Enhanced Catalytic CO Oxidation by Atomically Dispersed Pt1/Pt2 on Raised Graphene Oxide Platform
Chuanyi Jia,† Yujin Zhang,† Xijun Wang,† Wenhui Zhong, Oleg V. Prezhdo, Yi Luo, Jun Jiang*
J. Mater. Chem. A 2020, 8, 12485–12494
Abstract:
TheRevealing structure–activity relationships of graphene-supported atomically dispersed transition metal catalysts is of key importance in catalysis chemistry and materials science. Here we report a firstprinciples theoretical study on O2 activation and CO oxidation catalyzed by atomically dispersed Pt1/Pt2 anchored on a raised graphene (Gr) platform. The unique sharp-tip structure of the protuberant graphene platform (carbon-tip) can collect extra polarization electrons on the Pt-tip, promoting electron transfer between the catalyst and adsorbates, and greatly enhancing the chemical activity. Higher carbon-tips on defective graphene oxide with a carbon vacancy (Gr-V) produce more polarization electrons, a more localized electric field, and a larger up-shift of the d-orbital center of the Pt-tip, resulting in better catalytic performance. The carbon-tip enhancement reduces the energy barrier for the CO oxidation on Pt1O2/Gr-V from 1.19 eV to 0.56 eV compared with Pt1 on planar Gr. In addition, CO poisoning is significantly alleviated, with the CO poisoning rate Gq reduced from 2.00 eV to 0.52 eV. Moreover, a linear relationship between the activation barrier and the binding energy of adsorbates is found for various atomically dispersed Pt-tip catalysts on the raised graphene oxide platform. Importantly, the order of catalytic activity is consistent with the carbon-tip enhancement, i.e., Pt1O2/Gr-V > Pt2O4/Gr-V > Pt1O2/Gr > Pt2O4/Gr. These findings provide important guidance for rational design of atomically dispersed catalysts.

Substituted SrFeO3 as Robust Oxygen Sorbents for Thermochemical Air Separation: Correlating Redox Performance with Compositional and Structural Properties
Emily Krzystowczyk,† Xijun Wang,† Jian Dou, Vasudev Haribal, Fanxing Li*
Phys. Chem. Chem. Phys. 2020, 22, 8924–8932
Abstract:
Thermochemical air separation via cyclic redox reactions of oxide-based oxygen sorbents has the potential to achieve high energy efficiency. Although a number of promising sorbents have been investigated, further improvements in sorbent performance through a fundamental understanding of the structure–performance relationships are highly desirable. In this study, we systematically investigated the effects of A and B site dopants on the oxygen uptake/release properties (i.e., vacancy formation energy, reduction enthalpy, oxygen release temperature, and oxygen capacity) of the SrFeO3 family of perovskites as oxygen sorbents. A monotonic correlation between DFT calculated oxygen vacancy formation energy and oxygen release temperature demonstrates the effectiveness of DFT for guiding sorbent selection. Combining vacancy formation energy with stability analysis, dopants such as Ba and Mn have been identified for tuning the redox property of SrFeO3 sorbents, and increasing the oxygen capacity for temperature and pressure swings when compared to undoped SrFeO3. The Mn doped sample proved to be highly stable, with less than a 3% decrease in capacity over 1000 cycles. Although the dynamic nature of the redox process makes it difficult to use a single vacancy formation energy as the descriptor, a systematic approach was developed to correlate the oxygen storage capacities with the sorbents’ compositional properties and vacancy formation energies. The combination of DFT calculations with experimental studies from this study provides a potentially effective strategy for developing improved sorbents for thermochemical air separation.

Ohmic Contact Formation Mechanisms of TiN film on 4H-SiC
Zhongtao Wang,† Xijun Wang,† Wei Liu, Xiaoliang Ji, Chunqing Wang*
Ceram. Int. 2020, 46, 6, 7142–7148
Abstract:
The atomic structure, interfacial charge distribution, bonding nature, and interfacial electronic states of a 4H–SiC/TiN interface are systematically investigated to understand the Ohmic contact formation mechanisms of TiN to 4H–SiC. The experiment results clearly demonstrate that the well-arranged TiN (111)-oriented lattice planes are parallel to the (0001) SiC-oriented substrate, which is in line with the XRD results. In addition, the interface is coherent without any secondary phase layers, amorphous layers, or transition regions, which confirms the direct contact of TiN to SiC at the atomic scale, exhibiting a linear current–voltage relationship. Quantitatively, first-principle calculations reveal that the Schottky barrier height (SBH) is as low as 0.03 eV and that the band gap nearly vanishes at the interface, indicating an excellent Ohmic contact of TiN to 4H–SiC. Furthermore, the SBH is significantly reduced through the interfacial charge polarization effect and strong coupling of interfacial electronic states, enhancing the quantum electron transport. The present results provide insight into the complicated electronic effects of the Ohmic contact interface and indicate that TiN is a promising SiC Ohmic contact material for advanced next-generation power device applications.

Protecting the Nanoscale Properties of Ag Nanowires with an Epitaxially Grown Single SnO2 Monolayer as Corrosion Inhibitor
Yang Zhao,† Xijun Wang,† Shize Yang, Elisabeth Kuttner, Aidan A. Taylor, Reza Salemmilani, Xin Liu, Martin Moskovits, Binghui Wu, Ahmad Dehestani, Jian-Feng Li, Matthew F. Chisholm, Zhong-Qun Tian, Feng-Ru Fan*, Jun Jiang*, Galen D. Stucky*
J. Am. Chem. Soc. 2019, 141, 35, 13977–13986
Abstract:
The chemical reactivity and/or the diffusion of Ag atoms or ions during thermal processing can cause irreversible structural damage, hindering the application of Ag nanowires (NWs) in transparent conducting films and other applications that make use of the material’s nanoscale properties. Here, we describe a simple and effective method for growing monolayer SnO2 on the surface of Ag nanowires under ambient conditions, which protects the Ag nanowires from chemical and structural damage. Our results show that Sn2+ and Ag atoms undergo a redox reaction in the presence of water. First-principle simulations suggest a reasonable mechanism for SnO2 formation, showing that the interfacial polarization of the silver by the SnO2 can significantly reduce the affinity of Ag to O2, thereby greatly reducing the oxidation of the silver. The corresponding values (for example, before coating: 17.2 Ω/sq at 86.4%, after coating: 19.0 Ω/sq at 86.6%) show that the deposition of monolayer SnO2 enables the preservation of high transparency and conductivity of Ag. In sharp contrast to the large-scale degradation of pure Ag-NW films including the significant reduction of its electrical conductivity when subjected to a series of harsh corrosion environments, monolayer SnO2 coated Ag-NW films survive structurally and retain their electrical conductivity. Consequently, the thermal, electrical, and chemical stability properties we report here, and the simplicity of the technology used to achieve them, are among the very best reported for transparent conductor materials to date.

Modified Ceria for “Low‐Temperature” CO2 Utilization: A Chemical Looping Route to Exploit Industrial Waste Heat
Vasudev Pralhad Haribal,† Xijun Wang,†, Ryan Dudek, Courtney Paulus, Brian Turk, Raghubir Gupta, Fanxing Li*
Adv. Energy Mater. 2019, 9, 41, 1901963
Abstract:
Efficient CO2 utilization is key to limit global climate change. Carbon monoxide, which is a crucial feedstock for chemical synthesis, can be produced by splitting CO2. However, existing thermochemical routes are energy intensive requiring high operating temperatures. A hybrid redox process (HRP) involving CO2-to-CO conversion using a lattice oxygen-deprived redox catalyst at relatively low temperatures (

Catalytic Chemistry Predicted by a Charge Polarization Descriptor: Synergistic O2 Activation and CO Oxidation by Au−Cu Bimetallic Clusters on TiO2(101)
Chuanyi Jia,† Xijun Wang,† Wenhui Zhong, Zhunzhun Wang, Oleg V. Prezhdo, Yi Luo, Jun Jiang*
ACS Appl. Mater. Inter. 2019, 11, 9, 9629-9640
Abstract:
The versatile properties of bimetallic nanoparticles greatly expand the range of catalyzed chemical reactions. We demonstrate that surface chemistry can be understood and predicted using a simple adsorbate–surface interaction descriptor that relates charge polarization to chemical reactivity. Our density functional theory studies of O2 activation and CO oxidation catalyzed by Au7–Cu1 bimetallic nanoparticles supported on the TiO2(101) surface demonstrate that the generated oxidized Cu atom (CuOx) can efficiently inhibit the aggregation of the active Cu sites. Moreover, because of the strong dipole–dipole interaction between the surface and the adsorbate on the oxidized Cu site, the adsorption of CO + O2/CO + O can be significantly enhanced, which can decrease the CO oxidation barriers and further improve catalytic performance. The product of the two electric dipole moments provides a parameter that allows us to predict the key catalytic properties for different adsorption sites and reaction pathways. The reported findings provide important insights into the mechanism of chemical reactivity of metallic clusters and generate a valuable principle for catalyst design.

The “healing” effect of reduced graphene oxide in achieving robust room-temperature dilute ferromagnetism in oxygen deficient titanium dioxide
Qing Zhu,† Xijun Wang,† Jun Jiang*, An-Wu Xu*
J. Phys. Chem. C 2017, 121, 41, 22806–22814
Abstract:
Titanium dioxide (TiO2) is an important wide-band-gap semiconductor with promising application for next-generation spintronics. Unfortunately, the lack of inherent spin ordering enormously hinders the widening scope of TiO2, and the origination of ferromagnetic properties still needs to be comprehensively explored due to the fact that manipulating the magnetic property in semiconductor through defect engineering remains a great challenge. Here we systematically investigate the room-temperature ferromagnetism (RTFM) behavior of defective anatase TiO2–x with the exposed (001) facet grown on reduced graphene oxide (rGO). First-principles simulations were performed to examine two types of intrinsic oxygen defects in TiO2–x: vacancy on surface (VO-Sur) and at subsurface (VO-Sub), among which only the VO-Sub contributes a considerable magnetism. Interestingly, simulations revealed a so-called “healing” effect for the oxygen functional groups in rGO, by removing the VO-Sur defect of TiO2–x, which helps establish good interface and thereby ensures good coupling between rGO and VO-Sub defects. Calculations show that the interaction of rGO with Ti3+–oxygen vacancy associates alters spin asymmetric electron distribution around VO-Sub and consequently introduces significant spin asymmetric defect states near the Fermi level. Hence rGO triggers a significant magnetic enhancement in TiO2–x. Importantly, our findings pave a new avenue for effective design and manipulation of spin states in an undoped dilute ferromagnetic semiconductor for spintronics application.

Trimetallic TriStar Nanostructures: Tuning Electronic and Surface Structures for Enhanced Electrocatalytic Hydrogen Evolution
Nana Du,† Chengming Wang,† Xijun Wang,† Yue Lin, Jun Jiang, Yujie Xiong*
Adv. Mater. 2016, 28, 2077–2084
Abstract:
PtFeCo alloy nanostructures in a TriStar shape with tunable Fe and Co content are developed for the electrocatalytic hydrogen evolution reaction (HER). With electronic and surface structures well‐tailored, the PtFeCo nanostructures exhibit dramatically enhanced performance in HER against commercial Pt/C and other Pt‐based nanoparticles.

The Dynamic Phase Transition Modulation of Ion‐Liquid Gating VO2 Thin Film: Formation, Diffusion, and Recovery of Oxygen Vacancies
Shi Chen,† Xijun Wang,† Lele Fan, Guangming Liao, Yuliang Chen, Wangsheng Chu, Li Song, Jun Jiang,* Chongwen Zou*
Adv. Funct. Mater. 2016, 26, 3532-3541
Abstract:
Electrolyte gating with ionic liquids (IL) on correlated vanadium dioxide (VO2) nanowires/beams is effective to modulate the metal‐insulator transition (MIT) behavior. While for macrosize VO2 film, the gating treatment shows different phase modulation process and the intrinsic mechanism is still not clear, though the oxygen‐vacancy diffusion channel is always adopted for the explanation. Herein, the dynamic phase modulation of electrolyte gated VO2 films is investigated and the oxygen vacancies formation, diffusion, and recovery at the IL/oxide interface are observed. As a relatively slow electrochemical reaction, the gating effect gradually permeates from surface to the inside of VO2 film, along with an unsynchronized changes of integral electric, optical, and structure properties. First‐principles‐based theoretical calculation reveals that the oxygen vacancies can not only cause the structural deformations in monoclinic VO2, but also account for the MIT transition by inducing polarization charges and thereby adjusting the d‐orbital occupancy. The findings not only clarify the oxygen vacancies statement of electrolyte gated VO2 film, but also can be extended to other ionic liquid/oxide systems for better understanding of the surface electrochemical stability and electronic properties modulation.

Aggregation-Induced Intersystem Crossing: A Novel Strategy for Efficient Molecular Phosphorescence
Li Yang,† Xijun Wang,† Guozhen Zhang, Xiaofeng Chen, Guoqing Zhang, Jun Jiang*
Nanoscale 2016, 8, 17422–17426
Abstract:
“Aggregation-caused quenching” (ACQ) and “aggregation-induced emission” (AIE) are two well-known mechanisms for polymer luminescence. Here we proposed an alternative mechanism termed “aggregation-induced intersystem crossing” (AI-ISC). By aggregating certain fluorescent dye molecules, one can improve the energy matches between excited singlet and triplet states so as to promote the intersystem crossing (ISC) rate, and consequently prolong the lifetime of excited electrons by steering them into triplet states. First-principles calculations suggested that the enhanced ISC rate could substantially promote molecular phosphorescence in aggregated systems of originally fluorescent dye molecules, as later validated by experimental measurement. Meanwhile, the emission spectra experience a red shift along with the aggregation, providing a convenient knob to tune the phosphorescence wavelength. The proposed AI-ISC mechanism may open up a new design approach for the emerging luminescent material applications.
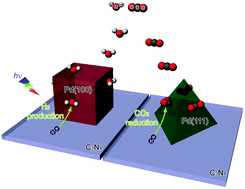
Two-dimensional g-C3N4: an ideal platform for examining facet selectivity of metal co-catalysts in photocatalysis
Song Bai,† Xijun Wang,† Canyu Hu, Maolin Xie, Jun Jiang*, Yujie Xiong*
Chem. Commun. 2014, 50, 6094–6097
Abstract:
Two-dimensional g-C3N4 nanosheets with few-layer thickness, ensuring equivalent charge migrations to various Pd facets, provide an ideal model system for reliably examining the facet selectivity of Pd co-catalysts. It reveals that reduction of CO2 can occur better on Pd{111} facets while H2O prefers to generate H2 on Pd{100}.

Multifunctional Fluorescent Probe for Sequential Detections of Glutathione and Caspase-3 in Vitro and in Cells
Rui Huang,† Xijun Wang,† Dingli Wang, Fang Liu, Bin Mei, Anming Tang, Jun Jiang,* Gaolin Liang*
Anaal. Chem. 2013, 85, 13, 6203–6207
Abstract:
Herein, we report a new “On–On” strategy based on the assembly and disassembly of fluorescein isothiocyanate nanoparticles (FITC-NPs) for sequential detections of glutathione (GSH) and caspase-3 (Casp3) with a multifunctional fluorescent probe 1. Theoretical investigations revealed the underlying mechanism that satisfactorily explained experimental results of such consecutive enhancements of fluorescence. Using this probe, we also successfully imaged the Casp3 activity in apoptotic cells.

Atomic Scale Analysis of the Enhanced Electro- and Photo-Catalytic Activity in High-Index Faceted Porous NiO Nanowires
Meng Shen,† Ali Han,† Xijun Wang,† Yun Goo Ro, Alireza Kargar, Yue Lin, Hua Guo, Pingwu Du,* Jun Jiang, Jingyu Zhang, Shadi A. Dayeh, Bin Xiang*
Sci. Rep. 2015, 5, 17, 8557
Abstract:
Catalysts play a significant role in clean renewable hydrogen fuel generation through water splitting reaction as the surface of most semiconductors proper for water splitting has poor performance for hydrogen gas evolution. The catalytic performance strongly depends on the atomic arrangement at the surface, which necessitates the correlation of the surface structure to the catalytic activity in well-controlled catalyst surfaces. Herein, we report a novel catalytic performance of simple-synthesized porous NiO nanowires (NWs) as catalyst/co-catalyst for the hydrogen evolution reaction (HER). The correlation of catalytic activity and atomic/surface structure is investigated by detailed high resolution transmission electron microscopy (HRTEM) exhibiting a strong dependence of NiO NW photo- and electrocatalytic HER performance on the density of exposed high-index-facet (HIF) atoms, which corroborates with theoretical calculations. Significantly, the optimized porous NiO NWs offer long-term electrocatalytic stability of over one day and 45 times higher photocatalytic hydrogen production compared to commercial NiO nanoparticles. Our results open new perspectives in the search for the development of structurally stable and chemically active semiconductor-based catalysts for cost-effective and efficient hydrogen fuel production at large scale.